O médico referido como Síndrome de Roberts uma malformação hereditária autossômica recessiva séria. Síndrome de Roberts às vezes é chamada Síndrome de Appelt-Gerken-Lenz, Síndrome de pseudotalidomida e também Roberts SC focomelia designadas. Esses nomes não descrevem diferentes estágios ou formas, mas são baseados principalmente nos descobridores da síndrome.
O que é a síndrome de Roberts?

© bluebackimage - stock.adobe.com
Como Síndrome de Roberts descreve uma malformação que ocorre muito raramente devido à determinação genética ou mutação. A ausência das quatro extremidades é particularmente característica. Via de regra, as previsões são muito ruins; em muitos casos, o desenvolvimento físico ou mental é severamente restringido. A maioria das pessoas afetadas morre no parto.
No final, os portadores da Síndrome de Roberts também são uma reminiscência de vítimas de Contergan porque têm deformidades e malformações semelhantes. Não há tratamento causal ou medidas preventivas para prevenir a síndrome de Roberts. Os médicos que diagnosticam a síndrome de Roberts - com base em um teste genético - procuram principalmente aliviar os sintomas e melhorar a qualidade de vida das pessoas afetadas. A síndrome de Roberts foi descrita pela primeira vez em 1919.
O cirurgião americano John Bingham Roberts escreveu a primeira descrição científica. No entanto, demorou 67 anos para que mais conhecimento surgisse. Foram Hans Appelt, Widukind Lenz e Hartmut Gerken, três geneticistas da Alemanha, que realizaram sua primeira pesquisa em 1966. Por esse motivo, a malformação hereditária é chamada de síndrome de Roberts ou também é conhecida como síndrome de Appelt-Gerken-Lenz.
causas
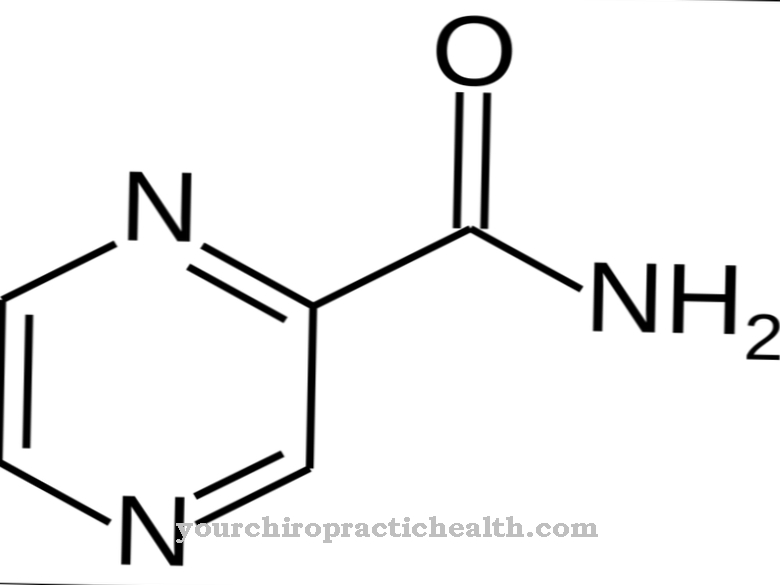
A síndrome de Roberts surge devido a mutações no gene ESCO2. O gene ESCO2 está localizado no locus do gene p21.1 do cromossomo 8. Acima de tudo, o produto do gene ESCO2 desempenha um papel essencial; em última análise, é a chamada N-acetiltransferase, que nos humanos é composta de exatamente 601 aminoácidos.
Quando a fase S entra e a divisão celular começa, as cromátides dobram. Essa abordagem desencadeia a síndrome. Até agora, no entanto, a frequência portadora da mutação é desconhecida. O gene ESCO2 consiste em um total de onze exons com 30,3 kb; não há como dizer qual frequência portadora está presente. As razões para a mutação não são conhecidas.
Sintomas, doenças e sinais
As pessoas afetadas que sofrem da síndrome de Roberts ou não nascem natimortas ou morrem logo após o nascimento apresentam vários sintomas que indicam a mutação correspondente. Há retardo mental, microcefalia (a cabeça é pequena), focomelia (o chamado "membro em selo") e também braquicefalia (cabeça curta ou redonda).
Em quase todos os casos, pode-se observar fenda labial e palatina; Também há hiperplasia do clitóris e do pênis (aumento). A córnea das pessoas afetadas fica turva ou o médico também pode detectar deformidades em órgãos internos (coração ou rins).
Diagnóstico e curso da doença
O médico faz um diagnóstico suspeito no início. Ele pode fazer isso sem problemas - com base nos sintomas. O diagnóstico só pode ser confirmado depois que a mutação for detectada por meio de um teste genético. Até que haja evidência genética de que realmente existe uma mutação responsável pela síndrome de Roberts, uma suspeita de diagnóstico é feita, não importa o quão claros sejam os sintomas da pessoa.
Em muitos casos, as pessoas afetadas morrem após o nascimento ou já nasceram como natimortos. Mas também há casos individuais em que o desenvolvimento mental normal foi documentado. No entanto, deve ser mencionado que esta é a exceção absoluta. O curso da doença e os prognósticos são em sua maioria negativos.
As medidas terapêuticas podem teoricamente reduzir o curso da doença ou favorecer os sintomas, mas também aqui não podemos falar de curso ou prognóstico positivo.
Complicações
Os que sofrem da síndrome de Robert geralmente morrem ao nascer ou logo depois. Se a criança doente sobreviver, quase sempre sofrerá graves danos mentais e físicos. A ausência dos quatro membros e outras anormalidades típicas estão associadas a uma dor considerável para a criança. A deficiência intelectual está associada a uma variedade de complicações - desde distúrbios do desenvolvimento a doenças secundárias específicas e exclusão social.
Em geral, a pessoa afetada sofre de várias queixas e seus efeitos tardios. Isso tem um impacto no estado mental da criança e dos pais. Se houver desenvolvimento mental normal, a pessoa afetada ainda precisará de apoio para o resto da vida. Outras complicações dependem dos sintomas específicos.
A falta de membros está associada ao repouso no leito e suas consequências típicas, enquanto a fenda labiopalatina causa distúrbios da fala. Outras complicações podem surgir ao tratar as queixas individuais. Infecções e lesões nervosas costumam ocorrer durante a cirurgia.
Devido às más condições físicas das pessoas afetadas, as queixas cardiovasculares e os distúrbios na cicatrização de feridas não podem ser excluídos. Se a medicação for administrada, efeitos colaterais e interações podem ocorrer ou reações alérgicas ocorrer.
Quando você deve ir ao médico?
As crianças que sofrem da síndrome de Roberts requerem tratamento médico rigoroso. Os diversos distúrbios físicos e mentais são tratados de forma reconstrutiva e cosmética, a fim de melhorar o bem-estar e a qualidade de vida. A fisioterapia geralmente é necessária para compensar quaisquer malformações nas mãos e pés. Os pais das crianças afetadas devem conversar com o médico sobre as medidas terapêuticas necessárias e consultar o respetivo especialista. Dependendo dos sintomas, ortopedistas, neurologistas, cirurgiões, oftalmologistas e dermatologistas, entre outros, estão envolvidos no tratamento.
O pediatra pode assumir as verificações regulares de acompanhamento, desde que a criança não precise ser tratada como paciente internada. Os pais e parentes da criança afetada geralmente também precisam de suporte terapêutico. Se a criança nascer morta ou se a doença for fatal, isso representa um fardo particularmente grande para os pais, que devem procurar um psicólogo adequado desde o início para trabalhar e superar o trauma. Como a síndrome de Roberts é uma doença genética, o aconselhamento genético é útil se você quiser ter filhos novamente.
Terapia e Tratamento
Não há terapia causal na síndrome de Roberts. Isso significa que principalmente os sintomas são tratados para que a qualidade de vida da pessoa afetada possa ser melhorada tanto quanto possível. No entanto, deve ser mencionado que em muitos casos as malformações são tão drásticas que a ajuda só pode ser dada em pequena medida.
O médico deve avaliar por si mesmo o quanto o tratamento pode ajudar. A síndrome de Roberts só precisa ser avaliada individualmente para que se possa decidir qual tratamento pode ser feito de vez em quando.
Acima de tudo, existem medidas corretivas para que seja alcançada uma melhoria na qualidade de vida. O médico decide sobre as correções cirúrgicas, que são principalmente estéticas e funcionais por natureza. Isso dá a oportunidade de aumentar a qualidade de vida da pessoa afetada. Uma das correções sem problemas é o tratamento da fenda labiopalatina.
Às vezes, as chamadas intervenções de cirurgia da mão podem tornar mais fácil segurar ou agarrar objetos. Visto que as malformações dos órgãos internos são características da síndrome de Roberts, aqui são realizados tratamentos individuais. O médico fica atento à extensão das malformações e deformidades dos órgãos, para que aqui também sejam realizadas terapias individuais.
O médico também deve tomar a decisão, se a síndrome de Roberts for tão pronunciada que a pessoa afetada às vezes tenha apenas algumas horas ou dias de vida, de não iniciar nenhum tratamento adicional. No final das contas, o médico espera apenas a morte natural do paciente.
prevenção
A síndrome de Roberts não pode ser evitada. Isso porque também não se sabe por que ocorre a mutação ou se existem fatores favoráveis.
Cuidados posteriores
O acompanhamento para a síndrome de Roberts deve ocorrer individualmente para cada pessoa afetada, pois os sintomas podem ser muito diferentes. No caso de malformações graves, o paciente não pode ser ajudado, muitas vezes morre durante o parto ou logo após. Os cuidados posteriores incluem tratamento medicamentoso ou cuidados paliativos.
Quando a cirurgia é necessária, as medidas mais importantes tomadas são bons cuidados com a ferida e controle da cicatriz cirúrgica. Isso geralmente é acompanhado por fisioterapia. Os cuidados de acompanhamento também podem incluir exames adicionais por especialistas, dependendo dos sintomas, que podem variar de opacidade da córnea a malformações dos órgãos internos.
No entanto, o acompanhamento independente geralmente não ocorre, pois a síndrome de Roberts é uma condição crônica e as pessoas afetadas devem ser tratadas por toda a vida. O especialista responsável deve decidir em conjunto com os familiares quais as opções de tratamento possíveis a longo prazo. Psicólogos também podem ter que ser consultados para isso, pois a doença pode representar uma grande carga emocional para os envolvidos. Os pais da criança, em particular, precisam de apoio emocional, pois na maioria dos casos os pacientes morrem logo após o nascimento ou são natimortos.
Você pode fazer isso sozinho
A síndrome de Robert pode aparecer em diferentes formas, que podem assumir diferentes cursos. Dependendo do curso, uma terapia individual deve ser elaborada em conjunto com o médico, que também inclui medidas de autoajuda para os familiares.
No caso de uma doença leve, os afetados primeiro precisam de várias operações e suporte fisioterapêutico. Na maioria das vezes, há malformações nos quatro membros, razão pela qual o uso de dispositivos auxiliares como muletas ou cadeira de rodas é necessário em todos os casos. Os familiares devem entrar em contato com um centro de apoio desde cedo para receber apoio no dia a dia. Como o sofrimento representa uma carga emocional considerável para os familiares, o tratamento terapêutico também é útil para os pais.
Após a cirurgia, a criança deve ser monitorada constantemente. O risco de complicações aumenta, mesmo após a internação. Os pais também devem entrar em contato com escolas especiais e jardins de infância especiais. Quanto mais cedo essas medidas forem iniciadas, menos estressante pode ser a vida de uma criança doente. Crianças com Síndrome de Roberts podem fazer fisioterapia em casa. Em qualquer caso, devem levar um estilo de vida saudável, com exercícios suficientes e uma dieta adequada.
.jpg)
.jpg)



.jpg)




.jpg)
.jpg)
.jpg)


.jpg)