o Síndrome de Bardet-Biedl, Além disso Síndrome de Laurence-Moon-Biedl-Bardet (LMBBS), é uma doença do campo das ciliopatias que ocorre exclusivamente por hereditariedade. A síndrome se manifesta na forma de múltiplas malformações que são desencadeadas por mudanças (mutações) em diferentes localizações de genes ou cromossomos.
O que é a síndrome de Bardet-Biedl?

© Creativa Images - stock.adobe.com
O quadro clínico definido pelos médicos Moon e Laurence e posteriormente por Bardet e Biedl é uma doença em que a distrofia retiniana ocorre como uma característica clinicamente significativa em combinação com outros sintomas. Devido a esta complicada situação médica inicial, a determinação final da doença BBS é difícil. Este quadro clínico foi registrado clinicamente pela primeira vez em 1866.
Quatro pessoas examinadas tinham retinite pigmentosa (distrofia retiniana, RP) em conjunto com paraplegia (paralisia espástica), bem como hipogenitalismo (órgãos genitais subdesenvolvidos) e deficiência mental. Em 1920, o médico francês Bardet descreveu uma doença composta por RP (distrofia retiniana), hipogenitalismo, polidactilia e obesidade.
O patologista de Praga, Biedl, também encontrou debilidade (confusão mental). Em 1925, os pesquisadores Weiss e Solis-Cohen resumiram os casos conhecidos e descreveram o quadro clínico como Síndrome de Laurence-Moon-Biedl-Bardet.
causas
Nos anos que se seguiram, a literatura médica apontou cada vez mais que os casos registrados por Laurence e Moon são uma forma especial rara que só ocorre em casos isolados junto com a BBS. Resultados de pesquisas médicas mais recentes atribuem a síndrome de Bardet-Biedl à área das ciliopatias (doenças ciliares).
Essas doenças apresentam um mau funcionamento comum dos chamados cílios (pequenos processos, antenas), que ocorrem na maioria das células do organismo humano. As ciliopatias são caracterizadas por transições fluidas e sobreposições entre diferentes doenças ciliares.
Sintomas, doenças e sinais
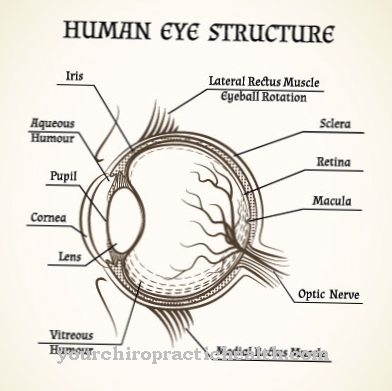
A principal característica da distrofia retiniana hereditária é um termo genérico que descreve o início da perda de função e a subsequente degeneração (destruição) dos fotorreceptores. Eles levam a uma perda progressiva (progressiva) da função visual. Os distúrbios visuais de rápida progressão geralmente aparecem muito cedo nas crianças, entre quatro e dez anos de idade. Eles se fazem sentir de maneiras diferentes, dependendo dos fotorreceptores afetados.
Como uma "forma de cone de haste" com curso característico de retinite pigmentosa (RP), a doença tem sua origem na periferia da retina (retina externa) e evolui para degeneração macular (destruição da visão aguçada) por meio de uma perda progressiva do campo de visão.
Com a obesidade (obesidade), o corpo mostra um acúmulo patológico de tecido adiposo. No caso da BBS, o aumento anormal do acúmulo de gordura nas pernas, estômago, nádegas, braços, tórax e quadris ocorre predominantemente como obesidade do tronco, sendo o tronco, as pernas e as coxas particularmente afetados. A polidactilia é um sintoma perceptível e uma característica significativa da síndrome de Bardet-Biedl. O achado não é fácil porque a polidactilia rudimentar é corrigida cirurgicamente após o nascimento.
Os raios X são capazes de fornecer mais informações. A polidactilia pode aparecer com sinais diferentes, por exemplo, como um dedo do pé rudimentar ou apêndice do dedo. Um dedo do pé ou dedo do pé pode ser formado adicionalmente ou apenas parcialmente. A hexadactilia unilateral no pé e / ou mão tem uma ligação adicional, a hexadactilia bilateral ocorre em ambos os pés e / ou mãos.
Dedos ou dedos que cresceram juntos (sindactilia) e encurtamento de um ou mais dedos do pé ou dedos (braquidactilia) também são sinais de BBS. Apenas alguns pacientes têm todas as quatro extremidades afetadas. O atraso do desenvolvimento mental é diferente. Apenas um pequeno número de pessoas afetadas apresenta retardo mental grave. Uma inteligência normalmente treinada é possível.
As crianças aprendem a falar e a andar até tarde e, às vezes, apresentam problemas de comportamento, como transtornos de ansiedade. Comportamentos compulsivos ou autistas, um baixo limiar de frustração e emocionalidade instável são outros efeitos colaterais possíveis. O familiar é o preferido, mas as alterações são rejeitadas. Anormalidades nos órgãos genitais internos e externos são comuns.
Outras alterações são hipospádia (a abertura uretral está acima ou abaixo, em vez de na parte frontal do pênis), abdômen ou testículos inguinais, constrições uretrais, constrição do prepúcio e válvulas uretrais posteriores. Em pacientes do sexo feminino, são conhecidas atresia vaginal (a vagina não é aberta), aberturas uretrais ausentes e lábios internos reduzidos.
Não é incomum que as mulheres afetadas tenham ciclos menstruais irregulares. As alterações renais são efeitos colaterais comuns. O achado depende do exame do trato urinário inferior e dos rins por meio de ultrassom (ultrassonografia).
Diagnóstico e curso da doença
A síndrome de Bardet-Biedl (BBS) tem seis sintomas principais, mas eles não ocorrem juntos em todos os casos. Os médicos presumem um achado correspondente se pelo menos quatro dos sintomas principais estiverem presentes. Como alternativa, existe uma grande probabilidade de que a doença esteja presente se o paciente tiver três sintomas principais e dois sintomas secundários.
Os seis principais sintomas são distrofia retinal, obesidade (acúmulo anormal de tecido adiposo, excesso de peso), polidactilia (excesso de dedos e / ou dedos), retardo mental (retardo do desenvolvimento mental), hipogenitalismo (órgãos genitais subdesenvolvidos) e doença renal. Os sintomas secundários de baixa frequência incluem atrasos na fala, déficits na fala, malformações cardíacas, ataxia (coordenação motora prejudicada), asma, diabetes mellitus (diabetes), doença de Crohn (inflamação do intestino grosso e / ou delgado), displasia das costelas e vértebras e cifoescoliose (anemia vertebral) em.
Complicações
Na síndrome de Laurence-Moon-Biedl-Bardet, as pessoas afetadas geralmente sofrem de perda da função visual. A perda não ocorre repentinamente, mas gradualmente. Na pior das hipóteses, as pessoas afetadas ficarão completamente cegas, o que geralmente não pode mais ser tratado.
Especialmente em jovens e crianças, a cegueira pode levar a graves queixas psicológicas ou até depressão. Os pacientes estão claramente limitados em sua vida cotidiana e sofrem de um campo de visão muito reduzido. Em muitos casos, a síndrome de Laurence-Moon-Biedl-Bardet também leva a problemas comportamentais, de modo que as crianças em particular podem sofrer bullying ou provocação.
O desenvolvimento das crianças também é significativamente retardado e restringido pela síndrome. Os transtornos de ansiedade também podem ocorrer. Não é incomum que a síndrome de Laurence-Moon-Biedl-Bardet leve a queixas psicológicas e depressão em parentes ou pais. Infelizmente, não é possível um tratamento causal da síndrome de Laurence-Moon-Biedl-Bardet.
Algumas reclamações podem ser limitadas. No entanto, um curso totalmente positivo da doença não se instala. A síndrome não reduz a expectativa de vida do paciente. Em alguns casos, as pessoas afetadas às vezes precisam da ajuda de outras pessoas em suas vidas diárias.
Quando você deve ir ao médico?
Como a síndrome de Laurence-Moon-Biedl-Bardet é uma doença hereditária, o diagnóstico pode ser feito no útero. O mais tardar após o nascimento, um médico deve ser consultado se sintomas típicos, como distúrbios visuais ou obesidade, forem observados. As malformações dos dedos dos pés e das mãos também são um indicador claro de uma doença.Os pais que perceberem os sintomas em seus filhos devem informar o pediatra imediatamente.
Um exame abrangente fornece informações sobre a doença. Depois disso, a terapia geralmente é iniciada diretamente, que consiste em vários tratamentos por ortopedistas, neurologistas, oftalmologistas, internistas e terapeutas, bem como fisioterapeutas. Outras visitas ao médico são necessárias se o tratamento não surtir o efeito desejado. O aconselhamento médico também é necessário em situações de emergência, por exemplo, se a criança cair devido a uma malformação ou tiver uma convulsão repentina. Se o doente apresentar sinais de desconforto emocional, os pais devem consultar um terapeuta adequado. As crianças mais velhas podem contactar o psicólogo escolar juntamente com os seus pais e discutir as medidas adequadas.
Terapia e Tratamento
Esta doença ocorre com base na herança autossômica recessiva, o que significa que ambas as cópias (alelos) de um gene BBS mostram uma alteração (mutação). Os pais do paciente são “mestiços” e cada um carrega um alelo modificado e um alelo inalterado do gene correspondente. Eles não têm a doença. As crianças só ficam doentes se o pai e a mãe transmitirem o alelo mutado. Com filhos adicionais, a probabilidade de repetição é de 25 por cento.
Uma opção de terapia causal ainda não é conhecida, uma vez que certos sintomas da doença ainda não podem ser atribuídos de forma conclusiva às várias alterações genéticas. Os sintomas e suas manifestações aparecem de forma diferente, mesmo em irmãos doentes. Uma vez que o quadro completo característico da BBS está presente apenas em casos raros, especialmente em crianças pequenas, um diagnóstico correspondente é difícil.
Devido aos sintomas oligossintomáticos frequentemente presentes, com os quais ocorrem muito poucos sintomas atípicos e apenas ligeiramente pronunciados, outros possíveis quadros clínicos devem ser considerados no diagnóstico diferencial. Alterações no mesmo gene podem levar a diferentes quadros clínicos, por exemplo, a síndrome de Joubert, Bardet-Biedl ou Meckel-Gruber.
Outlook e previsão
O prognóstico para a presença da síndrome de Laurence-Moon-Biedl-Bardet é geralmente ruim porque as malformações múltiplas são congênitas e incuráveis. Se quatro dos seis sintomas principais ocorrerem, o diagnóstico de síndrome de Laurence-Moon-Biedl-Bardet é confirmado. Numerosos sintomas secundários se somam aos sintomas principais. Isso inclui uma cegueira crescente.
É devido à complexidade dos sintomas que não há perspectiva de cura. Existe apenas uma chance medíocre de alívio perceptível dos sintomas. O número de possíveis malformações e distúrbios na síndrome de Laurence-Moon-Biedl-Bardet é tão grande que a doença hereditária é difícil de tratar. Em qualquer caso, o curso desta doença genética não pode ser influenciado. No entanto, os sintomas presentes podem ser parcialmente aliviados.
No entanto, o mau prognóstico geral não reduz a expectativa de vida das pessoas afetadas. Em idade avançada e depois de ficar cego, as pessoas afetadas podem ficar permanentemente dependentes de ajuda ou cuidados. Por meio de esforços médicos interdisciplinares, muitos portadores da síndrome de Laurence-Moon-Biedl-Bardet podem experimentar um curso um pouco mais brando da doença.
Os crescentes problemas visuais representam uma parte difícil e problemática da doença. Já ocorrem crescentes deficiências visuais nas crianças pequenas afetadas. Eles pioram com o tempo. Os problemas de visão não devem levar à cegueira em todas as pessoas afetadas. As sequelas psicológicas da síndrome de Laurence-Moon-Biedl-Bardet geralmente podem ser bem tratadas.
prevenção
A prevenção no sentido de prevenir esta doença não é possível. O monitoramento regular dos sintomas e dos sintomas associados é importante. Verificações repetidas da pressão arterial e função renal, aconselhamento nutricional, fisioterapia e terapia ocupacional, bem como terapia da fala são abordagens terapêuticas possíveis.
Cuidados posteriores
Na maioria dos casos, as pessoas afetadas com a síndrome de Laurence-Moon-Biedl-Bardet não têm opções especiais de acompanhamento disponíveis, de modo que um médico deve ser contatado e consultado desde o início nesta doença. Como regra, a autocura não pode ocorrer, então o tratamento por um médico é sempre necessário.
Uma vez que a síndrome de Laurence-Moon-Biedl-Bardet é uma doença hereditária, a pessoa em questão deve ser submetida a um exame genético e aconselhamento realizado se desejar ter filhos para que a síndrome de Laurence-Moon-Biedl-Bardet não passe para seus filhos é transmitido. Em muitos casos, os afetados dependem de intervenções cirúrgicas para aliviar as malformações e deformidades.
Aqui, a pessoa afetada deve descansar definitivamente após o procedimento e cuidar de seu corpo. O esforço ou outras atividades físicas e estressantes devem ser evitados em qualquer caso, a fim de não sobrecarregar desnecessariamente o corpo. Visto que a síndrome de Laurence-Moon-Biedl-Bardet também pode levar a um comportamento anormal, os pais devem apoiar e encorajar o desenvolvimento da criança. Também são necessárias discussões amorosas e intensas com a criança para prevenir transtornos psicológicos ou depressão.
Você pode fazer isso sozinho
A síndrome de Laurence-Moon-Biedl-Bardet tem vários sintomas, com o paciente frequentemente sofrendo mais de deficiência visual. Mesmo com crianças, a capacidade normal de ver começa a se deteriorar, de modo que são os pais que apresentam a criança ao médico e, assim, agilizam o diagnóstico. Desta forma, a doença pode ser tratada rapidamente, embora as opções de tratamento até agora tenham sido apenas de natureza sintomática.
Os distúrbios visuais aumentam cada vez mais nas crianças doentes e, portanto, prejudicam consideravelmente a vida cotidiana, fazendo com que a qualidade de vida dos afetados diminua. Pois os problemas de visão desenvolvem inúmeras dificuldades para o paciente na hora de frequentar a escola, no seu tempo livre e no que diz respeito à sua integridade física. O risco de acidentes também aumenta significativamente, por exemplo, no tráfego rodoviário. É por isso que os pais acompanham seus filhos doentes sempre que possível ou contratam pessoal de enfermagem para que o paciente não fique sozinho.
Em alguns casos, a doença se espalha ao ponto da cegueira. Uma vez que tal desenvolvimento já é evidente de antemão, os pacientes se preparam para ele. Os pais redesenham o espaço de convivência para que não contenha nenhuma fonte de perigo para o deficiente visual. Além disso, as vítimas cegas aprendem a usar um bastão comprido para que possam se mover independentemente fora de sua casa.
.jpg)
.jpg)



.jpg)




.jpg)
.jpg)
.jpg)


.jpg)