O muito raro Síndrome de Løken Sênior (SLSN) incorpora uma das muitas formas possíveis de ciliopatia hereditária. A síndrome hereditária autossômica recessiva é caracterizada pela associação de nefronoftise, uma forma especial de inflamação renal intersticial e degeneração retinal (distrofia retiniana). Dependendo do tipo de defeito genético, os sintomas de degeneração retinal aparecem durante ou logo após o nascimento ou mais tarde na infância.
O que é a síndrome de Løken sênior?

© SciePro - stock.adobe.com
o Síndrome de Løken Sênior (SLSN) representa uma das cerca de 20 formas conhecidas de ciliopatia. É diferenciada de outras formas de ciliopatia na ocorrência simultânea de nefronoftise (inflamação intersticial dos rins) e distrofia retinal com deficiência visual severa a extremamente severa.
Uma diferenciação exata do SLSN de outras ciliopatias nem sempre é possível porque há sobreposições genéticas e fenotípicas com outras ciliopatias. Os cílios desempenham um papel importante no movimento das células e no "transporte" extracelular de substâncias sólidas e líquidas, bem como em muitos processos sensoriais como os impulsos.
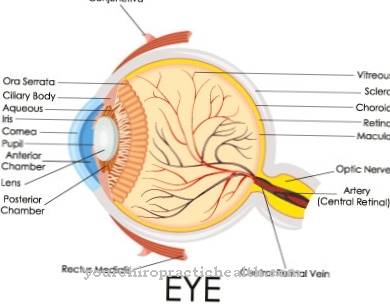
Cílios são protuberâncias citoplasmáticas da membrana celular. Ciliopatias, distúrbios funcionais dos cílios, têm, portanto, um efeito quase sistêmico em muitos processos metabólicos e no desempenho sensorial. A degeneração retiniana, que está associada à Síndrome de Løken Sênior, tem duas formas diferentes.
A amaurose de Leber é a forma mais grave, que já é perceptível durante e logo após o nascimento, devido a restrições do campo visual e reações pupilares enfraquecidas, bem como nistagmas.
causas
Como muitas outras ciliopatias, a Síndrome de Løken Sênior é causada exclusivamente por defeitos oligogênicos a poligênicos. No geral, o foco está em mutantes de sete genes diferentes em diferentes loci como gatilhos de SLNS. No futuro, outros genes poderão ser adicionados porque as pesquisas nesta área ainda não foram concluídas.
Os genes que codificam para proteínas nos cílios têm grande influência no desenvolvimento e, portanto, também na funcionalidade dos fotorreceptores da retina e das células do epitélio tubular dos rins. Uma vez que a doença é herdada de forma autossômica recessiva, ela só pode ocorrer se os defeitos genéticos estiverem presentes em ambos os cromossomos e colidirem.
Com a combinação de um gene “saudável” e um gene mutado, prevalece a codificação do gene não mutado, de forma que o SLNS não aparece, embora a predisposição genética seja latente.
Não há (ainda) um entendimento completo das conexões entre os defeitos genéticos que levam ao aparecimento da doença. Também estão em discussão as cadeias de interação epistática de certos genes, isto é, as interações de certos genes não homólogos uns com os outros.
Sintomas, doenças e sinais
Os sintomas de SLSN geralmente aparecem logo após o nascimento ou durante a fase de desenvolvimento da criança. Como os rins e a retina são igualmente afetados, os sinais e queixas se manifestam paralelamente em ambos os sistemas orgânicos, os rins e os olhos (retina).
Normalmente, os sinais de poliúria, excreção urinária anormalmente aumentada e a polidipsia associada, um aumento na ingestão de líquidos, já podem ser vistos logo após o nascimento. Os sinais de distrofia retiniana geralmente aparecem imediatamente após o nascimento na forma de restrições do campo visual e reflexos pupilares fracos.
Em muitos casos, nistagmas também são observados, em cascata, movimentos bruscos dos olhos que normalmente são desencadeados por impulsos dos canais semicirculares no ouvido interno ao acelerar. Os sintomas que aparecem nos rins e na retina são causados pelos processos degenerativos progressivos das células funcionais do epitélio renal e dos fotorreceptores da retina dos olhos.
Diagnóstico e curso da doença
Os sintomas que se desenvolvem em conexão com SLSN não são muito específicos e podem indicar uma série de outras doenças também. Portanto, exames abrangentes, como testes de função renal, exames laboratoriais de urina e um ultrassom da parte superior do abdome são recomendados para esclarecimento.
Se o exame for realizado na infância, um teste de função hepática também é recomendado para descartar fibrose hepática. No que diz respeito aos olhos, exames de fundo de olho, acuidade visual, campo de visão e visão colorida são usados para diferenciar de outras doenças. Um eletrorretinograma e a medição dos movimentos involuntários dos olhos completam os exames.
O curso do SLSN é determinado principalmente pelo progresso da degeneração do tecido renal funcional. Enquanto os processos de degradação na retina levam à cegueira, o prognóstico do curso geral da doença é muito desfavorável devido aos processos de degradação nos rins - se não for tratada.
Complicações
Por causa da síndrome de Senior Løken, as pessoas afetadas sofrem principalmente de problemas visuais graves. Via de regra, ocorrem imediatamente após o nascimento, de modo que os afetados já sofrem restrições significativas na vida cotidiana e, portanto, também no desenvolvimento na infância. Na pior das hipóteses, pode levar à cegueira completa.
Em muitos casos, os pacientes dependem da ajuda de outras pessoas em sua vida cotidiana e, especialmente em uma idade jovem, não conseguem fazer muitas coisas cotidianas facilmente. Isso também pode levar a queixas psicológicas ou depressão. Além disso, os sintomas desta síndrome também ocorrem nos rins, de modo que os pacientes podem sofrer de insuficiência renal. Na pior das hipóteses, isso pode levar à morte da pessoa em questão.
Os pacientes ficam então dependentes de um transplante de rim ou diálise. Como regra, a síndrome de Senior Løken sempre leva a uma perda total da visão, que não pode ser tratada. Os problemas renais podem ser limitados com um estilo de vida saudável de apoio. No entanto, na maioria dos casos, a expectativa de vida do paciente é significativamente reduzida pela síndrome.
Quando você deve ir ao médico?
A síndrome de Løken sênior deve sempre ser tratada por um médico. Na maioria dos casos, essa doença não pode curar a si mesma, de modo que a pessoa em questão está sempre dependente de tratamento médico. Visto que a síndrome de Senior Løken é uma doença genética, o aconselhamento genético também deve ser realizado se você quiser ter filhos, a fim de evitar que a síndrome seja transmitida aos seus descendentes. Um médico deve ser consultado se a pessoa em questão sentir desconforto nos olhos.
Esses sintomas aparecem sem nenhum motivo específico e não desaparecem por conta própria. Além disso, os sintomas renais também indicam síndrome de Senior Løken e devem ser examinados por um médico se ocorrerem juntamente com problemas oculares. Se a síndrome de Løken sênior não for tratada, na pior das hipóteses, a pessoa afetada pode ficar completamente cega. Portanto, a detecção precoce da síndrome é muito importante.
No caso da Síndrome de Løken Sênior, um clínico geral pode ser visto principalmente. O tratamento adicional é então realizado por um médico internista ou um oftalmologista.
Tratamento e Terapia
Se o diagnóstico de SLSN for positivo, é importante que as crianças afetadas sejam submetidas a testes regulares de função renal para serem capazes de reconhecer a progressão gradual da doença. Uma vez que não existe atualmente nenhuma substância medicinal conhecida que possa curar a nefronoftise juvenil, todos os métodos de tratamento visam inicialmente combater os sintomas e aliviar a dor.
Um suprimento controlado de sais é importante para manter o equilíbrio eletrolítico em equilíbrio, que pode mudar devido à perda constante de fluido. Se a progressão da doença atingiu um estágio crítico, a diálise de curto prazo e recorrente pode salvar vidas. A única terapia de longo prazo que pode ser considerada é o transplante de rim.
Também para o tratamento dos problemas retinais que se desenvolvem paralelamente aos problemas renais, não se conhece nenhum medicamento ou outro tratamento que possa retardar ou mesmo interromper a progressão da distrofia retiniana. O prognóstico para a progressão da distrofia retinal é, portanto, pobre. Em última análise, a doença sempre termina com uma perda total da visão, ou seja, com cegueira completa.
prevenção
A síndrome de Løken sênior deve-se exclusivamente a causas genéticas. Quaisquer medidas de precaução, como mudanças na dieta ou medicação preventiva, não seriam eficazes. Em princípio, um diagnóstico pré-natal é possível se os defeitos genéticos na família forem conhecidos e devidamente documentados.
Cuidados posteriores
A terapia para a síndrome de Løken na terceira idade vai direto para a fase de cuidados posteriores, durante a qual as crianças doentes devem ser examinadas regularmente.Os pais são responsáveis pelo cumprimento dos prazos de realização da prova de função renal. Eles devem seguir de perto as recomendações do médico para aliviar os sintomas e a dor de seus filhos.
A doença é hereditária, de modo que uma mudança na dieta apenas melhora as sensações corporais, mas não tem influência direta nos sintomas. No entanto, os pais devem garantir um bom equilíbrio eletrolítico e, para isso, tomar os sais prescritos pelo médico. Desta forma, o aumento da perda de fluido pode ser compensado.
Devem ser marcadas consultas regulares de diálise, dependendo da gravidade da doença. As crianças doentes freqüentemente se sentem debilitadas e precisam do apoio adequado. No contexto do planejamento familiar, o aconselhamento genético é útil se o defeito genético for conhecido. Este ponto também desempenha um papel relevante na prevenção e cuidados posteriores combinados.
Envolver familiares e amigos ajuda as crianças afetadas com os problemas cotidianos. Ao mesmo tempo, a segurança aumenta se o meio social for informado sobre a doença. Em conexão com o impacto negativo da doença na retina do olho, a psicoterapia pode ser útil.
Você pode fazer isso sozinho
As crianças que sofrem de Síndrome de Løken Sênior precisam de apoio particularmente amoroso na vida cotidiana. Acima de tudo, os pais devem certificar-se de que os rins são examinados regularmente quanto ao seu funcionamento. Isso permite que o progresso da doença seja identificado de forma confiável. Como parte do diagnóstico pré-natal, se o defeito genético for conhecido, um teste apropriado pode ser realizado.
No entanto, a doença hereditária não pode ser prevenida ou interrompida com uma dieta saudável ou certos medicamentos. Na vida cotidiana, é importante que os pacientes recebam sais controlados. Isso garante uma casa equilibrada de eletrólitos e evita que sejam eliminados devido à perda contínua de fluido. Em um estágio avançado da doença, podem surgir situações críticas. A diálise é então essencial. Freqüentemente, as crianças se acostumam com a diálise recorrente desde tenra idade. Mesmo assim, muitos pais esperam um transplante de rim para tornar a vida mais fácil para seus filhos.
Além dos problemas renais, o desenvolvimento negativo da retina do olho desempenha um papel importante. Infelizmente, nenhuma terapia medicamentosa bem-sucedida é conhecida até agora, de modo que a distrofia retiniana está progredindo. Devido ao mau prognóstico da doença, o apoio psicológico é recomendado para as famílias afetadas.








.jpg)


.jpg)