Existem 45 diferentes no total doenças de armazenamento lisossomal, que são um grupo heterogêneo de doenças metabólicas congênitas. Pessoas que sofrem de qualquer uma dessas doenças têm um defeito genético. Todas as doenças de armazenamento têm uma coisa em comum: uma determinada enzima está ausente ou apenas parcialmente funcional.
O que é doença de armazenamento lisossomal?

© designua - stock.adobe.com
Essas doenças congênitas de armazenamento são raras, pois menos de cinco em 10.000 pessoas são afetadas. As várias doenças têm um curso muito diferente e os sintomas podem variar amplamente.
As formas mais famosas de doença de armazenamento lisossomal são doença de Fabry, doença de Gaucher, doença de Pompe e mucopolissacaridose (MPS). Eles são freqüentemente chamados de "órfãos da medicina" porque o caminho para um diagnóstico específico e uma terapia adequada pode ser muito longo. Às vezes, pode levar anos para que as pessoas afetadas descubram o que está acontecendo com elas.
causas
As doenças de armazenamento lisossomal são caracterizadas por certas formas de doenças metabólicas hereditárias. O paciente carece de uma enzima importante que garante que o equilíbrio metabólico funcione sem problemas. Na forma menos pronunciada, esta enzima pelo menos não está presente em quantidades suficientes.
A tarefa das enzimas é eliminar poluentes e substâncias residuais que se acumulam no organismo humano por meio do metabolismo por meio dos lisossomas, ou processá-los novamente de forma que os sintomas não ocorram.
Se houver uma deficiência de enzima, esse ciclo de eliminação de funcionamento normal não é mais garantido. As substâncias nocivas se instalam nas células e interrompem o ciclo metabólico. Na fase inicial, as perturbações não têm um efeito perceptível, existem apenas algumas restrições. No entanto, se esse distúrbio metabólico não for tratado como resultado de uma deficiência enzimática, os sintomas se multiplicam porque as células ficam muito aumentadas.
Sintomas, doenças e sinais
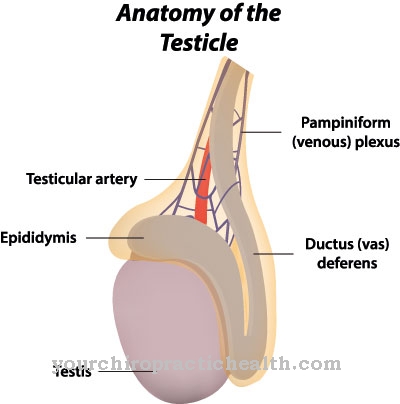
Na pior das hipóteses, eles afundam. As consequências são danos aos ossos, sistema nervoso, baço, rins, músculos ou coração. Devido à atividade enzimática reduzida ou ausente, a doença de Fabry faz com que a gordura (globotriaosilceramida, Gb3) seja armazenada nas células. Esses depósitos indesejados podem causar fortes dores nos dedos das mãos ou dos pés, derrame cerebral e danos aos rins.
Diagnóstico e curso da doença
Este quadro clínico afeta diferentes sistemas ao mesmo tempo: vasos sanguíneos, rins, coração e sistema nervoso. A doença de Gaucher hereditária autossômica recessiva causa uma mutação da enzima "beta-glucocerebrosidase" e leva a um acúmulo de substrato dentro das células, especialmente nos macrófagos (fagócitos) que pertencem ao sistema retículo-endotelial. O hemograma muda, o fígado e o baço aumentam de tamanho e os ossos doem.
A doença é progressiva e principalmente étnica, visto que ocorre na maioria dos casos em pessoas de ascendência judaica. A doença de Pompe também é conhecida como "deficiência de maltase ácida". O quadro clínico pertence ao grupo da glicogênese tipo 2. As pessoas afetadas não possuem a enzima "alfa-1,4-glicosidase" (maltase ácida) ou ela não está disponível em quantidades suficientes. Devido à degradação do glicogênio nos músculos, os pacientes sofrem com a destruição das células musculares na forma de armazenamento de açúcar.
A mucopolissacaridose tipo I (MPS), também conhecida como doença de Hunter, tem várias causas clínicas. A doença de Hurler é a forma mais grave e a doença de Scheie está no final da patogênese clínica. Existem transições de características diferentes entre essas duas formas de progressão. A propriedade mais notável é a degradação dos carboidratos que se acumulam nos lisossomas das células.
Os pacientes com doença de Hunter podem apresentar baixa estatura, baço e fígado aumentados, feições grosseiras, pele espessada, língua aumentada e dificuldade respiratória. Além disso, o esqueleto é frequentemente alterado na área da pélvis, coluna, ossos da mão e crânio. Hérnias umbilicais e [[hérnias inguinais] são possíveis.
Complicações
Na maioria dos casos, os sintomas ou complicações aparecem muito tarde nesta doença. Por isso, seu diagnóstico é tardio, impossibilitando o tratamento precoce na maioria dos casos. Sem tratamento, várias queixas e danos aos órgãos internos ocorrem à medida que a doença progride.
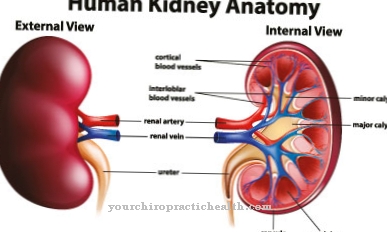
Os rins, fígado e baço são particularmente afetados. O coração também pode ser afetado por esta doença, que pode levar à morte cardíaca no pior dos casos. Além disso, ocorrem danos aos rins e as pessoas afetadas frequentemente sofrem de dores nos dedos dos pés ou das mãos. A paralisia também pode ocorrer se o cérebro tiver sido danificado por esta doença. O fígado e o baço podem aumentar de tamanho e causar fortes dores.
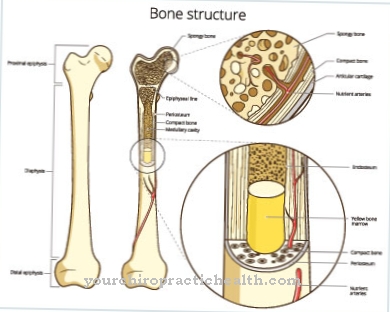
Não é incomum que os ossos da pessoa afetada sejam quebradiços e doloridos. O tratamento desta doença está se mostrando difícil. Em muitos casos, a expectativa de vida da pessoa afetada é significativamente reduzida. Geralmente, não há complicações específicas ao usar medicamentos. No entanto, um curso positivo da doença não pode ser garantido em todos os casos.
Você pode encontrar seu medicamento aqui
➔ Remédios para dorQuando você deve ir ao médico?
Queda de cabelo, problemas nas articulações e distúrbios de órgãos são possíveis sinais de doença de armazenamento lisossomal. Uma visita ao médico é recomendada se os sintomas continuarem recorrentes ou aparecerem repentinamente sem que uma causa seja encontrada. Se os sintomas estiverem relacionados a um defeito enzimático previamente diagnosticado ou outra doença grave, o médico responsável deve ser consultado. Uma doença de armazenamento não tratada pode levar à demência, infertilidade, neuropatias e outras complicações, algumas das quais são fatais. Portanto, todos os sintomas concebíveis devem ser examinados, mesmo que não haja suspeita específica.
Os sintomas da doença de armazenamento lisossomal podem aparecer em fases ou desenvolver-se insidiosamente, mas sempre requerem exame e tratamento. As pessoas afetadas devem falar diretamente com o médico de família ou com um internista. A terapia real geralmente ocorre em uma clínica especializada em doenças internas, embora a fisioterapia ou a psicoterapia possam ser conectadas dependendo dos sintomas. Em particular, medidas terapêuticas são indicadas devido ao curso muitas vezes negativo da doença.
Terapia e Tratamento
Dependendo de quão precocemente é feito um diagnóstico adequado, essas doenças hereditárias podem ser muito bem tratadas com terapia de reposição enzimática, de modo que as pessoas afetadas tenham muito menos queixas e, portanto, uma melhor qualidade de vida. Esta terapia de reposição é utilizada de acordo com o quadro clínico.
Pessoas que sofrem da doença de Gaucher não têm a “enzima ß-glucocerebrosidase”, que é produzida biotecnologicamente e infundida no corpo do paciente. Os lisossomos agem de forma eficiente e são capazes de absorver substâncias de seu ambiente imediato. Por este motivo, as enzimas utilizadas artificialmente são modificadas de forma a poderem ser fornecidas aos lisossomas de forma ideal.
Os macrófagos (fagócitos) decompõem os glicocerebrosídeos que se acumularam nas células. Essa terapia pode ser comparada à terapia com insulina para diabetes mellitus, com a diferença de que não é um hormônio ausente, mas uma enzima que não é fornecida. O corpo decompõe regularmente todas as substâncias, incluindo a enzima artificial fornecida.
Devido a esta degradação regular da substância, os pacientes têm de se submeter a esta terapia de infusão regularmente até o fim da vida. A terapia de reposição enzimática não atua sintomaticamente, mas combate diretamente a causa da doença hereditária. Os médicos chamam essa terapia de causal. Os princípios da terapia devem ser usados para todas as quatro doenças de armazenamento comuns acima mencionadas.
Os pacientes com Pompe também são tratados com terapia de infusão. Nesta doença, a enzima inexistente "alfa glucosidase ácida" é fornecida e ajuda a quebrar o glicogênio que se acumulou nos lisossomas dos músculos. Em pacientes com o tipo de doença “mucopolissacaridose tipo I”, a enzima lisossomal “alfa-iduronidase” não está presente ou não está presente em quantidades suficientes. É uma das doenças de armazenamento mais raras em que as moléculas de açúcar se acumulam em órgãos e tecidos.
Se o processo for normal, a enzima decompõe os mucopolissacarídeos. As moléculas de açúcar são de cadeia longa e estão envolvidas no desenvolvimento do tecido conjuntivo e de suporte, por exemplo, ossos, pele, fluidos articulares e cartilagem. Se o curso normal de degradação for perturbado devido à falta de enzima, os glicosaminoglicanos patológicos (GAG) acumulam-se nas células individuais. As opções de terapia futuras visam tomar comprimidos.
Outlook e previsão
O prognóstico para doenças de armazenamento é ruim. Descobriu-se que uma disposição genética é a causa do distúrbio de saúde. Requisitos legais proíbem médicos e cientistas de alterar a genética humana. Por esse motivo, a doença permanece por toda a vida e não tem perspectiva de recuperação.
O médico assistente concentra-se no tratamento dos sintomas que surgem. Se não forem tratadas, várias queixas aumentarão com o tempo. O sistema ósseo é danificado e surgem problemas nos órgãos. Na pior das hipóteses, os órgãos internos funcionarão mal e, por fim, sua função falhará. Isso ameaça a pessoa preocupada com a morte prematura.
O desafio da doença está no diagnóstico. Em um grande número de pacientes, queixas notáveis e fortemente perceptíveis ocorrem apenas mais tarde na vida. Como resultado, a doença genética permanece despercebida por muito tempo e o tratamento precoce da doença é difícil. Quanto mais tarde o diagnóstico for feito, mais desfavorável será o curso posterior. Em um estágio avançado da doença, os órgãos internos ou articulações já estão gravemente danificados. São necessárias intervenções cirúrgicas e, se a doença progredir de maneira desfavorável, apenas um órgão doador pode salvar a vida da pessoa afetada. O tratamento precoce é, portanto, essencial para um prognóstico melhor.
prevenção
Por se tratar de um defeito genético congênito que impede a expressão de uma enzima, essa doença não pode ser tratada preventivamente. No entanto, as mais recentes conquistas da engenharia genética podem fornecer uma abordagem neste campo.
Cuidados posteriores
Com esta doença, as pessoas sofrem de várias complicações e doenças diferentes. Regra geral, todos estes efeitos têm um efeito muito negativo na qualidade de vida da pessoa afectada, pelo que o diagnóstico deve ser feito desde muito cedo. Quanto mais cedo o médico for consultado, melhor geralmente será o curso posterior da doença.
A gravidade desta doença pode variar muito, de modo que muitas vezes não é possível fazer uma previsão geral. As pessoas afetadas sofrem graves danos aos órgãos internos. Os rins e o coração são os principais afetados, pelo que a criança pode morrer nos primeiros dias se os sintomas não forem corrigidos a tempo. Também existem depósitos de gordura em diferentes partes do corpo.
Os dedos das mãos e dos pés são particularmente afetados, o que pode levar a uma redução significativa da estética da pessoa afetada. Como regra, danos aos rins e ao cérebro ocorrem no curso posterior, de modo que a pessoa afetada morre como resultado desse dano. Os pais e parentes também costumam sofrer de depressão ou outros transtornos mentais devido à doença.
Você pode fazer isso sozinho
As doenças de depósito lisossomal muitas vezes requerem cuidados médicos intensivos. Freqüentemente, não há oportunidades suficientes para autoajuda. Os pais de crianças afetadas muitas vezes experimentam forte estresse em seu ambiente doméstico porque seus filhos precisam de cuidado e atenção constantes.
Os quadros clínicos das doenças de armazenamento individuais são diferentes. Existem formas fáceis e muito difíceis. Um exemplo é a doença de Gaucher. A ajuda dos pais muitas vezes se limita a alimentar a criança com deficiência grave. Em casos mais brandos, a expectativa de vida pode ser quase normal. No entanto, é necessária supervisão médica constante para evitar possíveis complicações. A atividade física regular é uma das terapias de acompanhamento que também pode ser realizada em casa. Além disso, um exame completo de rastreamento do câncer deve ser organizado. Isso requer visitas constantes ao médico com o filho dos pais. O mesmo se aplica a outras doenças de depósito lisossomal.
No caso de algumas doenças, além das deficiências físicas, também podem ocorrer deficiências mentais, que ainda requerem um suporte especial. Em formas mais brandas de certas doenças, como a doença de Hunter, inicialmente ocorrem apenas alterações esqueléticas e dismorfismo facial. Aqui, no entanto, o paciente afetado muitas vezes consegue levar uma vida independente. No entanto, exames médicos constantes também são necessários aqui, a fim de descartar possíveis complicações, como insuficiência cardíaca ou doenças respiratórias. O paciente pode lidar com o estresse psicológico causado por deformações físicas por meio de aconselhamento psicológico.


.jpg)


.jpg)




.jpg)

.jpg)

.jpg)
