Cobalaminas representam compostos químicos que pertencem ao grupo da vitamina B12. Eles ocorrem em todos os organismos. Eles são sintetizados apenas por bactérias.
O que são cobalaminas?
As cobalaminas são um grupo de compostos químicos com a mesma estrutura básica que pertencem ao complexo de vitamina B12. É um composto complexo com cobalto como átomo central. Eles são as únicas substâncias naturais que contêm cobalto conhecidas até hoje.
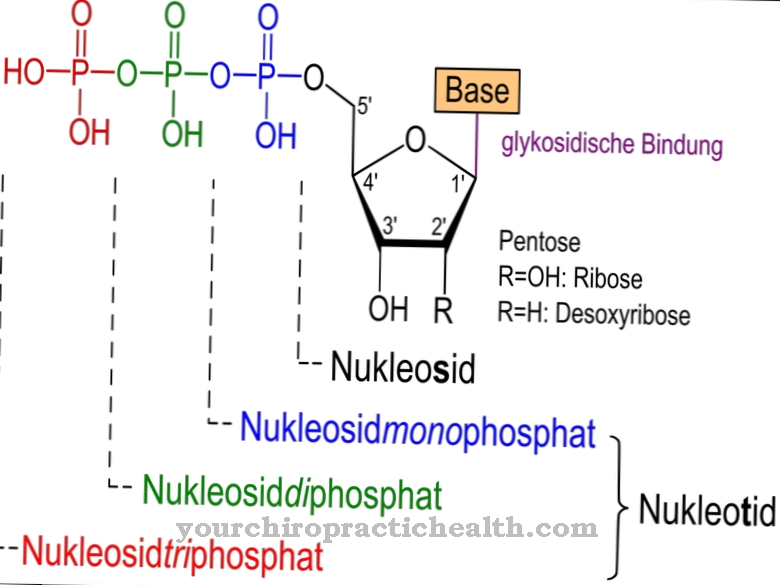
O átomo de cobalto é cercado por um total de seis ligantes. Quatro ligantes representam, cada um, um átomo de nitrogênio de um sistema de anel corrin planar.O quinto átomo de nitrogênio pertence a um anel 5,6-dimetilbenzimidazol, que está ligado ao anel corrin de uma maneira nucleotídica. Um sexto ligante é facilmente vinculado e intercambiável. Apenas este ligando intercambiável caracteriza o composto específico presente. A vitamina B12 real contém um resíduo de ciano como o sexto ligante e é, portanto, referida como cianocobalamina.
Outras cobalaminas importantes incluem aquacobalamina (vitamina B12a), hidroxicobalamina (vitamina B12b), nitritocobalamina (vitamina B12c), metilcobalamina (metil-B12, MeCbl) e, como uma coenzima extremamente importante, adenosilcobalamina (coenzima B12). Todos esses compostos também representam formas de armazenamento de vitamina B12. A cianocobalmina é a única vitamina B12 que pode ser usada na medicina. É imediatamente convertido em coenzima B12 no corpo.
Todas as formas de armazenamento do ingrediente ativo são absorvidas pelos alimentos. A vitamina B12 sintetizada por bactérias no intestino grosso não pode ser usada por humanos porque a absorção da cobalamina ocorre no intestino delgado.
Função, efeito e tarefas
A vitamina B12 desempenha funções importantes na formação do sangue, divisão celular e no sistema nervoso. Ele só participa de duas reações enzimáticas no organismo, mas são de importância biológica central.
A enzima N5-metil-tetrahidrofolato-homocisteína-S-metiltransferase (metionina sintase) funciona como um doador do grupo metil com a ajuda da coenzima B12. A metionina sintase reativa o transportador do grupo metil S-adenosilmetionina (SAM) por um lado e remetila a homocisteína em metionina, por outro. Se houver deficiência ou falha de vitamina B12, a homocisteína se acumula no sangue. O aumento das concentrações de homocisteína representa um fator de risco para a arteriosclerose.Além disso, a enzima N5-metil-THF também se acumula e, portanto, garante uma deficiência secundária de THF (ácido tetrahidrofólico).
O THF suporta a estrutura das bases de purina adenina e guanina, bem como a base de pirimidina, timina. As bases de nitrogênio estão envolvidas na construção dos ácidos nucléicos DNA e RNA. Se o THF estiver ausente, a síntese do ácido nucleico é interrompida. A segunda função da vitamina B12 é apoiar a enzima metilmalonil-CoA mutase. A metilmalonil-CoA mutase decompõe os ácidos graxos ímpares com a formação de propionil-CoAs. Propionil-CoAs é então introduzido no ciclo do ácido cítrico. Um metabólito neste processo é o metilmalonil-CoA. Se houver falta de vitamina B12, o metilmalonil-CoA se acumula, o que pode levar a sintomas neurológicos.
Educação, ocorrência, propriedades e valores ideais
As cobalaminas não podem ser produzidas no metabolismo vegetal, animal ou humano. Apenas algumas bactérias são capazes de sintetizar esse ingrediente ativo. Isso também inclui bactérias intestinais humanas.
Uma vez que a síntese da cobalamina em humanos ocorre no intestino grosso, mas a vitamina B12 é absorvida no intestino delgado com a ajuda do fator intrínseco, a cobalamina formada no intestino não pode ser usada. Os humanos dependem de suprimentos de comida. Ao mesmo tempo, há um processo bioquímico que permite que a vitamina B12 seja repetidamente transportada de volta ao intestino delgado por meio de ácidos biliares, onde é reabsorvida. Isso significa que, uma vez que o reservatório do fígado esteja cheio, ele durará vários anos, mesmo que não haja suprimento de vitamina B12. O fígado pode armazenar de 2.000 a 5.000 microgramas de cobalamina.
A necessidade diária mínima para adultos é de cerca de 3 microgramas. A necessidade de filhos é menor e aumenta com o tempo. Mulheres grávidas e amamentando têm uma necessidade maior, que é de 3,5 a 4 microgramas por dia. Após 450 a 750 dias, metade da cobalamina existente se esgota. Fontes importantes de cobalamina são o fígado e as entranhas de vários animais de fazenda, arenque, carne bovina, queijo, ovos de galinha e atum. A vitamina B12 está quase ausente nos alimentos vegetais. No caso de um estilo de vida vegetariano, é necessária suplementação adicional.
Doenças e distúrbios
Devido à grande importância das reações bioquímicas suportadas pelas cobalaminas, a deficiência de vitamina B12 leva a sérios problemas de saúde.
Uma deficiência pode resultar, por um lado, de uma dieta puramente vegetariana e, por outro lado, da perda do fator intrínseco. O fator intrínseco é uma glicoproteína que se liga à cobalamina no intestino delgado e, portanto, a redireciona para uso. Essa proteína é produzida nas células parietais do estômago. Em doenças gástricas com insuficiência das células parietais, um fator intrínseco não pode mais ser formado. Não é mais possível usar a cobalamina existente novamente. A falta de vitamina B12 inibe a metilação da homocisteína em metionina.
O nível de homocisteína no sangue aumenta e o risco de arteriosclerose aumenta. Ao mesmo tempo, N5-metil-tetrahidrofolato ((N5-metil-THF) se acumula. Ocorre deficiência de THF. Como resultado, a síntese de ácido nucleico não funciona mais corretamente. Processos com alta necessidade de ácido nucleico, como a formação de sangue, são inibidos. O número de células sanguíneas diminui, com os eritrócitos restantes aumentando de tamanho à medida que são preenchidos com hemoglobina.
O resultado é o que se conhece como anemia perniciosa (maligna), que não é causada por deficiência de ferro. Pode ser tratada com ácido fólico. A deficiência de cobalamina, entretanto, persiste e ainda causa queixas neurológicas, como mielose funicular ou polineuropatias, por meio da interrupção da metilmalonil-CoA mutase pelo acúmulo de ácido metilmalônico no plasma.


.jpg)


.jpg)


.jpg)

.jpg)

.jpg)
