Debaixo de Síndrome TAR, Inglês Síndrome de rádio ausente de trombocitopenia, a medicina entende uma síndrome de malformação, cujos principais sintomas incluem falha dos raios e trombocitopenia. Acredita-se que a causa da síndrome seja uma mutação genética hereditária. O tratamento nos primeiros anos de vida consiste principalmente na transfusão de plaquetas.
O que é a síndrome TAR?

© Reing - stock.adobe.com
o Síndrome TAR é um complexo de múltiplas malformações que se manifesta em recém-nascidos. Os principais sintomas da síndrome de malformação hereditária são falha bilateral do raio e falta de plaquetas sanguíneas. Por causa dos sintomas, a síndrome às vezes é datada Síndrome de aplasia-trombocitopenia radial o discurso. A síndrome TAR foi descrita em pouco mais de 100 casos. A prevalência exata não é conhecida, mas o complexo de sintomas é considerado relativamente raro.
A síndrome foi descrita pela primeira vez em 1929. Os americanos H. M. Greenwald e J. Sherman são considerados os primeiros a descrevê-la. De acordo com a documentação até agora, as mulheres têm mais probabilidade de ser afetadas por malformações do que os homens. Devido à sua raridade, a síndrome não foi totalmente pesquisada. A pesquisa de causa obteve sucessos parciais, mas até agora não foi capaz de fornecer uma explicação suficiente para todo o complexo.
causas
Em 2007, foi identificada uma possível causa para a síndrome TAR que corresponde a uma mutação genética. Os sintomas parciais do complexo são causados por uma microdeleção no cromossomo 1 no locus gênico q21.1. Neste contexto, estamos falando sobre a síndrome de deleção 1q21.1. O cromossomo 1 até agora foi associado a várias doenças hereditárias.
Mutações nesta localização do gene podem desencadear a síndrome de Usher, doença de Gaucher ou doença de Alzheimer, por exemplo. O cromossomo 1 está presente como um par de cromossomos em todas as células do corpo e corresponde ao maior cromossomo humano. A mutação associada à síndrome TAR parece necessariamente estar presente em todos os pacientes. No entanto, a mutação não explica adequadamente os sintomas individuais da síndrome.
A síndrome TAR é considerada uma doença hereditária. Acúmulo familiar foi observado nos casos documentados até o momento. A hereditariedade parece ser um modo autossômico recessivo de herança e uma variabilidade relativamente grande na manifestação.
Sintomas, doenças e sinais
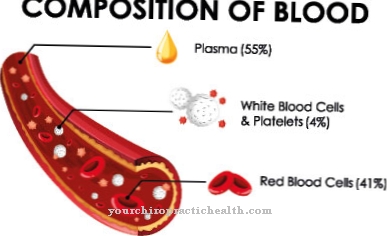
Todos os pacientes com síndrome TAR têm trombocitopenia. A falta de plaquetas leva a um aumento da tendência a sangrar. Eles diminuem principalmente nos primeiros dois anos de vida. Nos primeiros meses, pode ocorrer hemorragia intracraniana, que pode promover retardo motor ou mental. Bilateralmente, todos os pacientes com a síndrome também não têm raios.
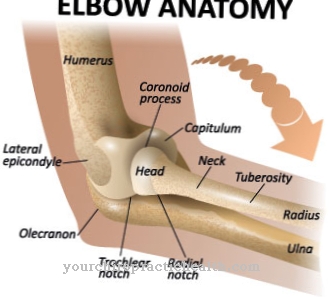
O polegar da pessoa afetada está lá, mas só funciona de forma anormal. Freqüentemente, há um desvio radial da mão, que se manifesta como uma deformidade da mão torta. A ulna de todos os pacientes com TAR é encurtada e parcialmente curvada. Cerca de um terço dos pacientes não tem o úmero, que geralmente também é encurtado e tem um efeito displásico. As articulações do cotovelo, ombro e mão são restritas em sua mobilidade.
Em alguns casos, também ocorrem alterações no sangue. Em dois terços dos casos, há leucócitos muito aumentados. Freqüentemente, há uma alergia ou intolerância ao leite de vaca que promove diarréia ou agrava a trombocitopenia. Em cerca de metade de todos os pacientes, os sintomas estão associados à displasia das extremidades inferiores.
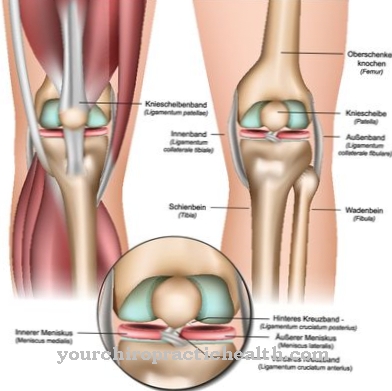
Em particular, displasia da anca, coxa valga, subluxação da articulação do joelho ou displasia patelar com luxação são sintomas comuns. O joelho pode ficar tenso. As posições dos pés e dedos dos pés geralmente são anormais. Muitos dos afetados também sofrem de baixa estatura ou um defeito cardíaco no sentido de uma tetralogia de Fallot ou um defeito do septo atrial. O olho geralmente apresenta ptose ou glaucoma.
Diagnóstico e curso da doença
Nos primeiros meses de vida, o médico observará uma tendência ao sangramento e trombocitopenia nos pacientes com TAR, que deve diferenciar da anemia de Fanconi no diagnóstico diferencial. Na imagem de raios-X, a síndrome TAR é particularmente evidente na falha dos raios em ambos os lados e nos desalinhamentos resultantes.
Em termos de diagnóstico diferencial, a síndrome de Holt-Oram e a síndrome de Roberts também devem ser consideradas. Assim que os primeiros dois anos de vida se passam, o prognóstico para pacientes com síndrome TAR é bastante favorável. Em casos individuais, o prognóstico depende dos sintomas associados, como o defeito cardíaco.
Complicações
Várias malformações ocorrem na síndrome TAR. Em primeiro lugar, as malformações levam a um aumento significativo da tendência a sangrar. As pessoas afetadas sofrem de sangramento intenso, mesmo com ferimentos muito leves e leves, que não podem ser interrompidos facilmente. O sangramento freqüentemente ocorre nas gengivas ou no nariz e tem um efeito muito negativo na qualidade de vida da pessoa em questão.
Além disso, pode ocorrer retardo mental devido à síndrome TAR. Em suas vidas, os pacientes muitas vezes dependem da ajuda de outras pessoas e não podem fazer muitas coisas do dia a dia sozinhos. A mobilidade dos ombros e das mãos também é significativamente restringida pela síndrome, pois o úmero está ausente. Além disso, pode causar um defeito cardíaco ou desconforto aos olhos.
A síndrome geralmente está associada à diminuição da expectativa de vida. Os pais ou parentes também sofrem frequentemente de problemas psicológicos ou depressão. O tratamento sintomático da síndrome TAR geralmente não envolve complicações. Infelizmente, nem todas as reclamações podem ser completamente restritas.
Quando você deve ir ao médico?
Na maioria dos casos, com a síndrome TAR, a pessoa afetada precisará de avaliação médica e tratamento. Não pode curar de forma independente, de modo que a pessoa afetada por esta doença depende sempre de um diagnóstico médico. Quanto mais cedo a síndrome for reconhecida, melhor será geralmente o curso posterior da doença. Por ser uma doença hereditária, nenhuma cura completa pode ocorrer. Se a pessoa afetada pela síndrome deseja ter filhos, o aconselhamento genético também pode ser usado.
No caso de síndrome TAR, um médico deve ser consultado se a pessoa em questão sofrer de retardo mental grave. Como regra, os pacientes dependem da ajuda de outras pessoas em suas vidas. A mobilidade da pessoa afetada também pode ser restringida pela síndrome TAR, sendo necessária uma consulta médica. Não é incomum que os órgãos internos sejam afetados por vários defeitos. O diagnóstico da síndrome TAR pode ser feito por um clínico geral ou pediatra. Para tratamento posterior, é necessária uma visita a um especialista.
Terapia e Tratamento
A síndrome TAR não pode ser tratada causalmente ou especificamente. Até agora, apenas tratamentos sintomáticos estão disponíveis. A correção das malformações não pode ocorrer nos primeiros anos de vida devido à tendência ao sangramento. Em fases posteriores da vida, as intervenções cirúrgicas reconstrutivas podem corrigir os raios ausentes e vários mal posicionamentos. Nos primeiros anos de vida é fundamental prevenir todo sangramento ou hemorragia.
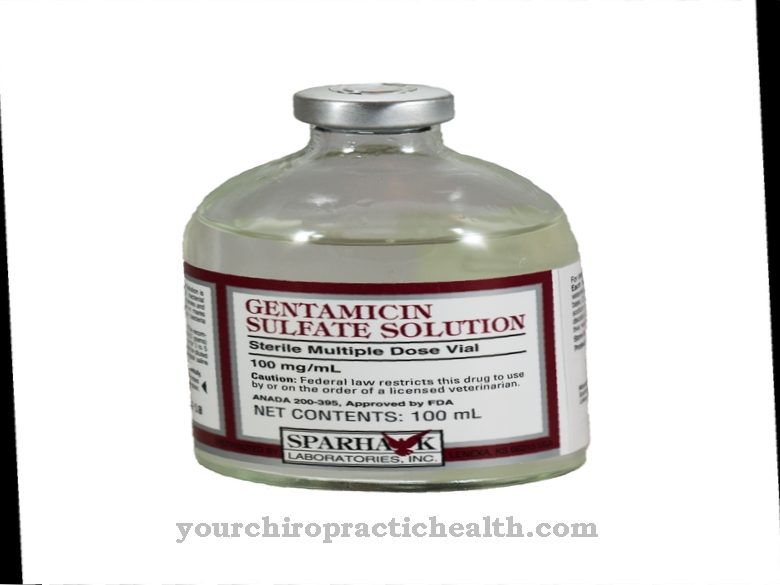
O objetivo da terapia inicial é, acima de tudo, reduzir as consequências significativas da doença. As trombocitopenias graves nos primeiros anos de vida exigem transfusões de plaquetas. Por si só, não há distúrbio do desenvolvimento motor. As limitações neurológicas também são raras. O desenvolvimento mental é imperceptível. Todos os retardos são, portanto, na melhor das hipóteses, uma consequência de sangramento intracraniano, que deve ser evitado com transfusões.
Quando a trombocitopenia cede, são realizadas as medidas de tratamento cirúrgico plástico. Essas medidas são acompanhadas por [[[etapas do tratamento fisioterapêutico]], que visam garantir o desenvolvimento motor perfeito. Na idade adulta, os pacientes geralmente não dependem mais de quaisquer medidas de tratamento e levam uma vida amplamente normal com uma qualidade de vida irrestrita.
Você pode encontrar seu medicamento aqui
➔ Medicamentos para tratamento de feridas e lesõesprevenção
As causas definitivas da síndrome TAR ainda não foram esclarecidas. Por isso, a síndrome ainda não pode ser prevenida. No entanto, tudo indica que fatores genéticos desempenham um papel na síndrome. Portanto, o aconselhamento genético para as pessoas afetadas pode ser amplamente descrito como uma medida preventiva.
Cuidados posteriores
Os cuidados de acompanhamento para a síndrome TAR dependem do tipo e da gravidade das malformações. Após um procedimento cirúrgico, que é uma opção para pequenas malformações, o paciente necessita de um acompanhamento extenso. Em caso de sangramento agudo, é necessário atendimento imediato na clínica. O paciente então precisa de alguns dias de descanso.
Um exame final de acompanhamento visa determinar as medidas de tratamento adicionais. Pessoas que sofrem de síndrome TAR devem consultar seu médico regularmente para esclarecer seu estado atual de saúde. As complicações médicas nem sempre são aparentes para o paciente, especialmente com sangramento interno. No caso de trombopenia grave, pode ser necessária uma transfusão de sangue, que é realizada na clínica e geralmente associada a um interrogatório.
O paciente, por sua vez, precisa de descanso e proteção. A hospitalização geralmente é indicada. Pacientes que sofrem de síndrome TAR precisam de um médico especialista. O médico responsável é geralmente o internista ou o clínico geral que já está envolvido no tratamento. A hospitalização de longo prazo faz sentido para queixas crônicas. O paciente também deve entrar em contato com um fisioterapeuta e outros especialistas. O paciente também pode precisar de suporte psicológico.
Você pode fazer isso sozinho
A síndrome TAR só pode ser tratada sintomaticamente. O paciente deve ficar atento aos sinais de alerta e informar o médico para que a transfusão de sangue seja realizada precocemente. Após essa transfusão, o corpo fica enfraquecido e é importante garantir uma dieta balanceada que apoie o corpo na produção de sangue.
Após o tratamento cirúrgico das malformações, aplicar repouso e repouso no leito. O paciente deve cuidar das feridas conforme orientação do médico para evitar inflamação e outras complicações. No caso de malformações nos membros, o tratamento fisioterapêutico também pode ser necessário. As pessoas afetadas podem fazer fisioterapia em casa e melhorar a coordenação dos membros afetados com exercícios regulares.
Caso essas medidas não obtenham o resultado desejado, o médico deve ser consultado. Como a síndrome TAR é uma doença extremamente rara, um médico especialista deve assumir a terapia. É aconselhável pesquisar fóruns na Internet para outras pessoas que sofrem, pois existem apenas alguns grupos de autoajuda para a doença. Por fim, é importante fazer as consultas necessárias ao médico para evitar complicações graves. A síndrome TAR deve ser monitorada de perto devido a qualquer transfusão de sangue.
.jpg)




.jpg)



.jpg)
.jpg)