A hipoxantina é um desvio de purina e vem na forma ligada como uma nucleobase e na forma livre, e. B. na urina. Também é encontrado nas glândulas e na medula óssea. Como um produto de desaminação da adenina, a hipoxantina é oxidada em ácido úrico e xantina. Com menos frequência, forma uma estrutura básica de ácidos nucléicos.
O que é hipoxantina guanina fosforibosiltransferase?
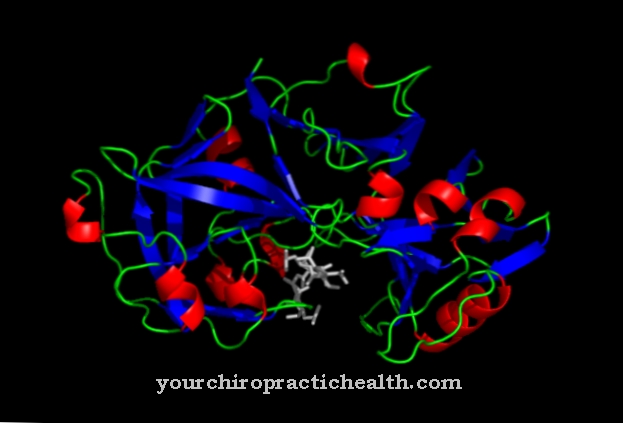
A enzima tetramérica é formada a partir de hipoxantina e guanina Hipoxantina guanina fosforibosil transferase.
Os tetrâmeros são macromoléculas que consistem em quatro blocos de construção semelhantes, mais precisamente de monômeros. A enzima é uma das mais importantes no metabolismo das purinas dos eucariotos, é sensível a alterações gênicas e pode causar desvios em humanos por meio de mutações gênicas, que se expressam em certas doenças metabólicas. Esses são z. B. Síndromes de Lesch-Nyhan e Kelley-Seegmiller.
Função, efeito e tarefas
A enzima hipoxantina-guanina-fosforibosiltransferase aumenta o metabolismo das purinas e sua eficácia energética.
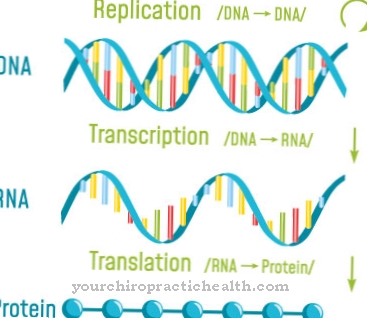
Isso é baseado em bases purinas, que são ácidos nucléicos que são estruturalmente derivados de purinas. São eles a xantina, a hipoxantina, a adenina e a guanina, que se ligam a outras bases por meio de ligações de hidrogênio. Essas ligações têm uma grande influência na dupla hélice e na replicação do DNA e desempenham um papel na biossíntese de proteínas.
As bases de purina podem ser recicladas por duas enzimas. Além da hipoxantina guanina fosforibosil transferase, esta é a adenina fosforibosil transferase. Ambos formam um nucleotídeo por meio de um resíduo de fosforibosila, que por sua vez é um bloco de construção básico dos ácidos nucléicos no DNA e no RNA. A molécula consiste em açúcar, base e fosfato e controla funções regulatórias vitais nas células. Durante o aumento, o ATP é salvo e a formação de ácido úrico é reduzida.
Se as bases de purinas forem recicladas, isso é conhecido como via de recuperação. Este é um termo geral para vias metabólicas nas quais a síntese de uma biomolécula surge a partir de produtos de degradação. O organismo realiza seu próprio processo de reciclagem, onde cerca de noventa por cento das bases purinas são reaproveitadas e dez por cento são excretadas. Isso mostra a eficiência da reciclagem de base de purina e a importância da hipoxantina-guanina fosforibosil transferase.
Educação, ocorrência, propriedades e valores ideais
Se ocorrerem mutações no gene HPRT, o tamanho e os aminoácidos podem mudar. Isso pode ser a incorporação de sequências de DNA adicionais ou nucleotídeos, que por sua vez leva a uma produção incorreta do produto do gene que é codificado no respectivo gene, ou mesmo à deleção de toda a sequência. É z. B. altera a sequência de aminoácidos, surgem doenças como a gota.
As doenças metabólicas, como a síndrome de Lesch-Nyhan, como resultado de um defeito genético, são particularmente graves. Isso é herdado de forma recessiva ligada ao X, o que significa que afeta principalmente homens que têm apenas um cromossomo X. O defeito genético pode estar presente em mulheres, mas só surge como doença quando os dois cromossomos X são afetados, o que é relativamente raro. Na maioria das vezes, o segundo cromossomo X compensa o defeito do primeiro.
Você pode encontrar seu medicamento aqui
➔ Medicamentos para a saúde da bexiga e do trato urinárioDoenças e distúrbios
A síndrome se manifesta por uma deficiência da hipoxantina guanina fosforibosil transferase. A enzima não é produzida devido ao defeito genético. Devido à mutação e à falta de reciclagem e conversão das bases de guanina e hipoxantina, há um acúmulo de bases de purinas que precisam ser acumuladas e excretadas pelo corpo.
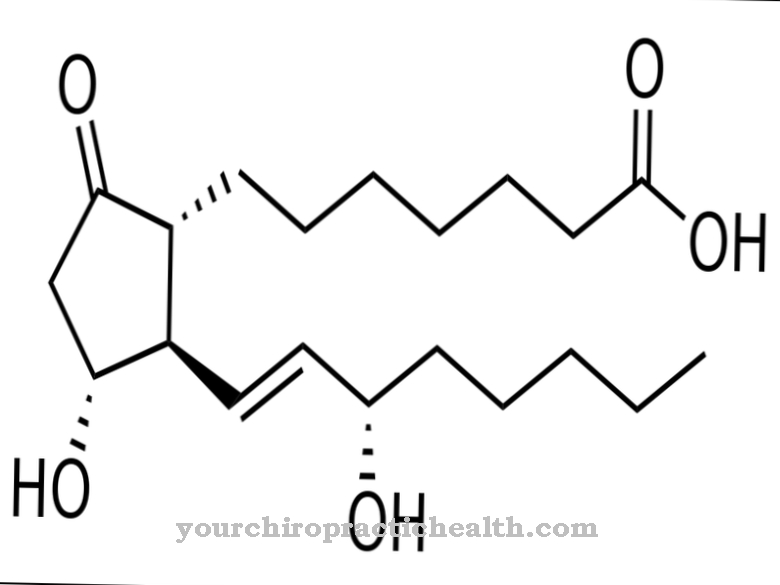
A degradação ocorre por meio do produto intermediário xantina, que é convertido em ácido úrico e excretado pelos rins. Se esse processo for restrito, cristais de ácido úrico se formarão na área das articulações, o que desencadeará mais ataques de gota. A enzima não é mais produzida, o nível de ácido úrico no tecido e no sangue aumenta e o sistema nervoso central é perturbado.
A síndrome de Lesch-Nyhan não é diretamente visível no nascimento. Uma posição perceptível das pernas e a tendência da criança de se mover pouco e se desenvolver mais lentamente só podem ser vistas após cerca de dez meses. A síndrome é fraca e grave. Uma secreção aumentada de ácido úrico e ataques de gota mais leves são as formas mais leves; se forem graves, podem ocorrer lesões autoprovocadas, deficiência mental grave e agressão. A automutilação ocorre por meio de mordidas nos dedos ou lábios. Ao morder as extremidades, muitas vezes pode-se observar que as pessoas afetadas limitam sua autoagressão a apenas uma mão. A agressão, por sua vez, costuma ser dirigida contra pessoas próximas a você, como irmãos ou pais.
A forma mais grave da doença é caracterizada por disfunções neurológicas múltiplas e uma tendência muito pronunciada para a automutilação. A síndrome se manifesta por espasticidade, distonia, hipotonia, coreoatetose e uma maior disposição para reagir aos reflexos. As qualidades mentais e o desenvolvimento são severamente restringidos. Nessa condição, a síndrome também pode levar à morte de forma particularmente drástica.
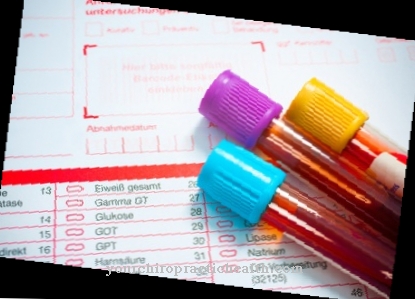
A doença é diagnosticada por meio de uma fotografia médica. O nível de ácido úrico na urina e no sangue é medido e a atividade da hipoxantina-guanina fosforibosiltransferase no tecido e no sangue. Este último é bastante reduzido e também pode estar presente no período pré-natal.
O tratamento da doença é difícil. A cura não é possível e sem tratamento a criança morre nos primeiros anos de vida. Em alguns casos, os dentes de leite precisam ser extraídos como medida preventiva. Outras abordagens terapêuticas incluem a redução dos níveis de ácido úrico usando drogas como o alopurinol, que atua como um inibidor da gota. As bases purinas não são recicladas, mas o ácido úrico é melhor decomposto. As respectivas doenças, infecções e lesões nervosas também são tratadas e aconselha-se uma dieta especial, que geralmente não contém carne e é pobre em purinas.
Também estão sendo realizadas pesquisas sobre os efeitos colaterais psicossomáticos da estimulação cerebral profunda. A medicina espera que isso impeça a agressão e a automutilação. A síndrome de Kelley-Seegmiller, por outro lado, é a forma mais branda de deficiência de hipoxantina-guanina fosforibosiltransferase. Também aqui é produzido ácido úrico em excesso e surgem doenças precoces de gota. Os primeiros indícios da síndrome são cristais laranja na fralda da criança, infecções do trato urinário e urolitíase. Gota ou artrite aguda se desenvolvem durante a puberdade.
O subdesenvolvimento mental e os ataques próprios, como ocorre com a síndrome de Lesch-Nyhan, não são o caso, no máximo podem levar a distúrbios de atenção. O tratamento precoce geralmente permite que as pessoas afetadas tenham uma expectativa de vida normal.

.jpg)


.jpg)


.jpg)

.jpg)

.jpg)
