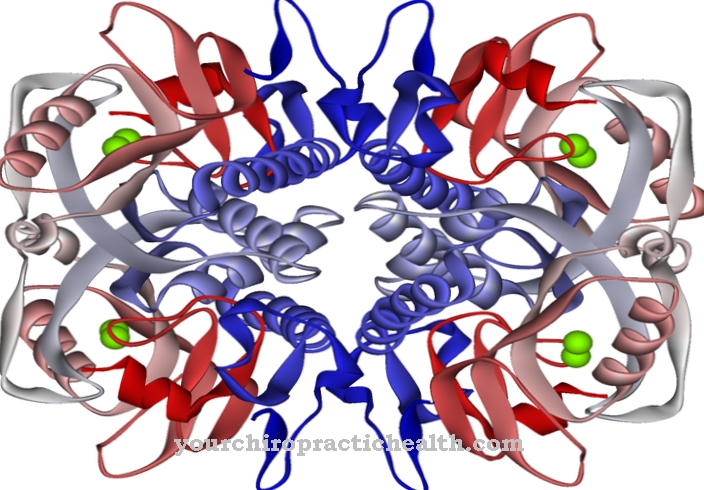
o Lipoproteína lipase (LPL) pertence às lipases e desempenha um papel crucial no metabolismo dos lipídios. É responsável por dividir os triglicerídeos nos quilomícrons e nas lipoproteínas de densidade muito baixa (VLDL) em ácidos graxos e monoacilglicerol. Os ácidos graxos liberados são usados para gerar energia ou aumentar a gordura corporal.
O que é lipase de lipoproteína?
A lipoproteína lipase (LPL) é uma enzima que é uma das lipases. As lipases são responsáveis por quebrar os triglicerídeos (triacilgliceróis) em ácidos graxos e glicerina. Os triglicerídeos são ésteres do álcool triplo glicerina, com três ácidos graxos cada, conhecidos como gorduras ou óleos graxos.
As gorduras dietéticas são absorvidas com os alimentos e, primeiro, decompostas pelas lipases extracelulares do pâncreas no intestino. No entanto, alguns triglicerídeos chegam à corrente sanguínea através do soro quando são absorvidos no intestino delgado, onde são ligados a lipoproteínas que garantem sua capacidade de transporte no sangue. A lipoproteína lipase é a enzima que decompõe os triglicerídeos ligados às lipoproteínas em ácidos graxos e monoacilglicerol. Consiste em 448 aminoácidos e é dependente da coenzima apolipoproteína C2 para sua função.
A lipoproteína lipase é uma enzima solúvel em água que se liga às células endoteliais dos vasos sanguíneos por meio de certas glicoproteínas (proteoglicanos). É produzido no fígado. A enzima catalisa a hidrólise dos triglicerídeos para formar duas moléculas de ácido graxo e uma molécula de monoacilglicerol. As apolipoproteínas são as moléculas transportadoras das triglicerinas e permitem o seu transporte em meio aquoso. A apolipoproteína C2 também atua como um receptor para a lipoproteína lipase e, portanto, ativa a hidrólise dos triglicerídeos.
Função, efeito e tarefas
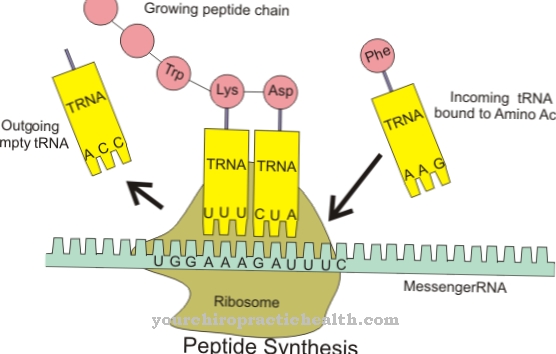
A função da lipase lipoproteica é catalisar completamente a degradação no sangue das gorduras absorvidas pelas células intestinais. Primeiro, as gorduras dietéticas são decompostas em ácidos graxos e glicerina pelas lipases pancreáticas no intestino delgado. Outros triglicerídeos entram no sangue por absorção através do intestino delgado e se ligam às lipoproteínas para formar um complexo lipídio-proteína.
Isso cria quilomícrons. Representam partículas de lipoproteínas com um diâmetro de 0,5 a 1 micrômetro e sua densidade é inferior a 1000 g / ml. O núcleo lipídico contém principalmente triglicerídeos com uma pequena quantidade de ésteres de colesterol. A casca dos quilomícrons contendo colesterol contém fosfolipídios como elemento estrutural. As apolipoproteínas às quais os triglicerídeos estão ligados agora também são armazenadas nesta casca. Os quilomícrons contêm 90% de triglicerídeos. Eles entram na corrente sanguínea a partir do intestino delgado através do sistema linfático. Os triglicerídeos são decompostos em ácidos graxos e glicerina com a ajuda da LPL, especialmente nos capilares do músculo e do tecido adiposo.
Os ácidos graxos são usados no tecido muscular para gerar energia ou no tecido adiposo para formar triglicerídeos endógenos como gordura de armazenamento. Após cerca de dez horas de abstinência alimentar, não é possível detectar mais quilomícrons no sangue porque os triglicerídeos são então completamente decompostos. Outros componentes do sangue são as chamadas VLDL (lipoproteína de densidade muito baixa). Essas unidades estruturais são liberadas do fígado e contêm triglicerídeos, fosfolipídios e colesterol. O VLDL transporta esses componentes através da corrente sanguínea do fígado para os órgãos individuais.
Dessa forma, os triglicerídeos são decompostos pela lipoproteína lipase e os ácidos graxos liberados são absorvidos pelas células do corpo. A diminuição dos triglicerídeos converte o VLDL em LDL (Lipoproteína de Baixa Densidade). O LDL contém principalmente fosfolipídios, ésteres de colesterol e lipoproteínas
Educação, ocorrência, propriedades e valores ideais
A lipoproteína lipase é sintetizada no fígado. Além das lipases pancreáticas, representa outra lipase extracelular.A LPL está localizada na parte externa das membranas das células endoteliais em uma ampla variedade de órgãos, incluindo células de gordura. Lá, ele é conectado às membranas celulares por meio dos chamados proteoglicanos.
No entanto, é de particular importância para as células endoteliais dos vasos sanguíneos, uma vez que aqui pode controlar diretamente a hidrólise dos triglicerídeos nos quilomícrons e VLDLs. A heparina é injetada para medir a atividade da lipoprotease. A heparina remove a ligação das lipases lipoproteicas dos proteoglicanos, de forma que após uma injeção de heparina ocorre um aumento da concentração de lipases lipoproteicas livres, que pode ser determinada por sua atividade. Este exame pode, entre outras coisas, determinar uma deficiência na lipase lipoproteica.
Doenças e distúrbios
A falta de lipase de lipoproteína freqüentemente leva a sérios problemas de saúde. Se houver pouca lipoproteína lipase ou se sua atividade for insuficiente devido a um defeito genético, os triglicerídeos nos quilomícrons e nas VLDLs podem ser mal decompostos ou não podem ser decompostos.
A deficiência de lipoproteína lipase pode ser principalmente genética, bem como secundária à quimioterapia, por exemplo. A deficiência primária de LPL é rara e é causada por um defeito genético autossômico recessivo. Desenvolve-se a chamada quilomicronemia, caracterizada por um soro leitoso e cremoso e é referida como hiperlipidemia do tipo I. Os triglicerídeos nos quilomícrons não são mais decompostos. Como resultado, pancreatidas graves com intolerância ao leite e dor abdominal ocorrem repetidamente.
Além disso, surtos de xantomas e hepatomegalia desenvolvem-se constantemente. As únicas opções de tratamento são uma dieta com baixo teor de gordura e sem álcool. Esta doença é freqüentemente causada por mutações no gene LPL no cromossomo 8 ou no gene APOC2. A forma secundária de hiperlipidemia do tipo I geralmente ocorre com a quimioterapia e é apenas de natureza temporária.
.jpg)

.jpg)


.jpg)


.jpg)

.jpg)

.jpg)
