o Terapia de reposição enzimática é usado para tratar doenças de armazenamento lisossomal, nas quais a falta de enzimas leva a um acúmulo patológico de produtos de degradação nos lisossomas das células.
As enzimas ausentes devido a defeitos genéticos são compensadas por infusões intravenosas regulares. Como as enzimas sintéticas infundidas não podem cruzar a barreira hematoencefálica devido ao seu tamanho molecular, a terapia só funciona para doenças de armazenamento lisossomal que não afetam o sistema nervoso central.
O que é terapia de reposição enzimática?

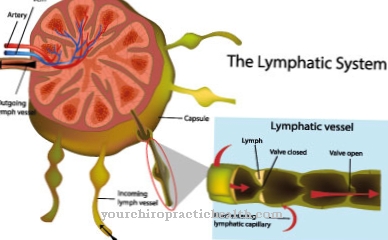
Os lisossomos são organelas celulares especiais nas quais substâncias estranhas e endógenas são decompostas e parcialmente recicladas. As enzimas hidrolisantes específicas são necessárias para a degradação e transporte das substâncias. São proteases, nucleases, lipases e substâncias transportadoras.
Uma série de defeitos genéticos conhecidos pode levar a uma falha de certas enzimas, de modo que alguns produtos de degradação se acumulam nos lisossomos em quantidades patológicas e se acumulam até atingirem a matriz extracelular, ou seja, os espaços intercelulares, de forma descontrolada. Todos os defeitos genéticos que levam à falha de pelo menos uma hidrolase necessária são resumidos sob o termo doença de armazenamento lisossomal. A terapia de reposição enzimática (ERT, terapia de reposição enzimática) é usado para substituir as enzimas endógenas em falta por enzimas produzidas sinteticamente.
Como as hidrolases são compostas de moléculas relativamente grandes, não podem ser absorvidas do intestino sem primeiro serem quebradas e inativadas, de modo que só podem ser administradas por infusão intravenosa. No entanto, o tamanho das moléculas de enzima também evita que a barreira hematoencefálica seja cruzada, de modo que a terapia só pode ser eficaz para doenças de armazenamento lisossomal que não afetam o sistema nervoso central (SNC).
Função, efeito e objetivos
Mais de 50 distúrbios metabólicos lisossomais diferentes são conhecidos, cada um dos quais pode ser atribuído a um defeito monogenético. As doenças de armazenamento lisossomal podem ser divididas em sete classes diferentes dependendo das substâncias excessivamente armazenadas devido ao defeito enzimático existente.
Mucopolissacaridoses e oligossacaridoses são principalmente adequadas para uma TRE. O objetivo da ERT é sempre compensar a deficiência enzimática específica por meio de enzimas fornecidas artificialmente, a fim de interromper a doença ou, pelo menos, ter um curso mais brando. Em detalhe, as enzimas de substituição estão disponíveis para as seguintes doenças de armazenamento lisossomal:
- Doença de Gaucher
- Doença de Pompe
- Doença de Fabry
- Síndrome de Hurler-Pfaundler (mucopolissacaridose I)
- Doença de Hunter (mucopolissacaridose II)
• Síndrome de Maroteaux-Lamy (mucopolissacaridose VI) • Niemann-Pick B
A doença de Gaucher é a doença de armazenamento lisossomal mais comum. Ocorre em três variantes diferentes, duas das quais também afetam o sistema nervoso. Na forma não neuropática, o baço é particularmente afetado, o que aumenta muito e causa danos secundários, como anemia e danos à medula óssea. Os sintomas típicos são dores nos ossos e nas articulações e distúrbios circulatórios. A variante neuropática aguda da doença mostra um curso grave e oferece pouca chance de sobrevivência além dos primeiros dois anos de vida.
A doença de armazenamento, doença de Pompe, é devida a uma deficiência da enzima alfa-1,4-glucosidase, que está envolvida em um grande número de processos metabólicos. A doença de Pompe causa um enorme aumento do coração (cardiomegalia) e insuficiência cardíaca. Existem cursos precoces e sérios que aparecem nos primeiros meses de vida, bem como formas mais brandas que só aparecem nos últimos anos de vida.
A doença de Fabry é causada por um defeito genético ligado ao X, portanto, apenas meninos e homens podem ser afetados pela doença de armazenamento. A doença geralmente leva a sintomas na infância avançada, incluindo ataques de dor, queratomas da pele, problemas renais e danos ao músculo cardíaco. A deficiência da enzima alfa-galactosidase A leva ao acúmulo de ceramida trihexoside, que é a causa do desencadeamento dos sintomas, que também podem afetar o sistema nervoso autônomo.
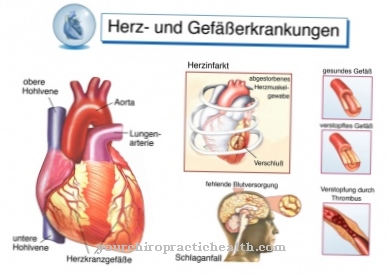
Não é incomum que os danos levem a um ataque cardíaco, infarto renal ou mesmo um derrame. A síndrome de Hurler-Pfaundler também é conhecida como mucopolissacaridose, tipo I e é causada por uma perturbação do metabolismo dos glicosaminoglicanos. A doença está associada a uma ampla variedade de sintomas, incluindo deficiência mental grave e alterações esqueléticas graves. O curso da doença é grave, de modo que a expectativa média de vida é de 11 a 14 anos. A doença de Hunter corresponde à mucopolissacaridose, tipo 2 e é - como a doença de Hurler - causada por um defeito ligado ao X. A doença é caracterizada por cursos de gravidade variável, desde a primeira infância até cursos leves que só aparecem em homens adultos.
Devido aos sintomas cardíacos mais comuns, como defeitos nas válvulas cardíacas e problemas no músculo cardíaco, a expectativa de vida varia de normal a ligeiramente restrita. A síndrome de Maroteaux-Lamy (MPS VI) é uma das mucopolissacaridoses herdadas como traço autossômico recessivo porque o defeito genético que a causa não está no cromossomo X. A doença é muito rara, com um caso a cada 455.000 nascimentos. São conhecidas as formas leves e graves.
Os sintomas são aumento do fígado e baço, síndrome do túnel do carpo e alterações nas válvulas cardíacas. O Niemann-Pick B é uma lipidose da esfingomielina, uma das doenças de armazenamento lisossomal causada por um defeito genético no cromossomo 11. Enquanto o tipo B da doença afeta principalmente o fígado e o baço, o tipo A também tem problemas neuronais consideráveis.
Você pode encontrar seu medicamento aqui
➔ Remédios para dorRiscos, efeitos colaterais e perigos
Uma vez que muitas das doenças de armazenamento lisossomal que podem ser tratadas com terapia de reposição enzimática seguem um curso severo com uma taxa de mortalidade correspondentemente mais alta se não forem tratadas, o maior risco na TRE é que a enzima de reposição selecionada não funciona ou funciona muito fracamente.
Outro risco reside menos na terapia em si do que no fato de a doença subjacente ser reconhecida tarde demais, de modo que a TRE pode parar no curso do curso, mas o dano que já foi causado não pode regredir. Aproximadamente cada segundo paciente tratado reage temporariamente às infusões com sintomas como febre e calafrios. As razões para isso ainda não são totalmente compreendidas. Alguns pacientes reagem formando anticorpos e há casos conhecidos em que os pacientes reagiram com erupções cutâneas e broncoespasmo.















.jpg)